Синдром Альпорта – наследственное заболевание почек, вызванное изменением синтеза коллагена типа IV, образующего базальные мембраны почечных клубочков, структуры внутреннего уха, хрусталика глаза. Мужчины страдают развернутой формой болезни с тяжелой симптоматикой. Женщины часто являются носителями гена, оставаясь здоровыми, или проявления болезни у них выражены слабо. Основные симптомы – микрогематурия, протеинурия, почечная недостаточность, сенсорная тугоухость, деформация и вывих хрусталика, катаракта. Диагноз устанавливается согласно клинико-анамнестическим данным, результатам общего анализа мочи, исследования биоптата почки, аудиометрии и офтальмологического осмотра. Лечение симптоматическое, включает терапию иАПФ и БРА.
Q87.8 Другие уточненные синдромы врожденных аномалий, не классифицированные в других рубриках
Семейные случаи гематурической нефропатии впервые привлекли внимание исследователей в 1902 году. Спустя почти 30 лет, в 1927 году американский врач А. Альпорт обнаружил частую сочетаемость гематурии с тугоухостью и уремией у мужчин, в то время как у женщин симптомы отсутствовали или были слабовыраженными.
Он предположил наследственный характер болезни, которая впоследствии была названа синдромом Альпорта. Синонимы – наследственный нефрит 1 типа, гематурический нефрит, семейный гломерулонефрит. Распространенность невысока – 1 случай на 5 тысяч человек. На долю патологии приходится 1% больных с почечной недостаточностью, 2,3% пациентов, перенесших трансплантацию почек.
Заболевание диагностируется у людей всех рас, но соотношение различных форм неодинаково.
Синдром Альпорта
По своей природе синдром является гетерогенным наследственным заболеванием – его развитие провоцируется дефектом генов, которые кодируют структуру различных цепей IV типа коллагена. Генетические изменения представлены делециями, сплайсинг, миссенс и нонсенс-мутациями. Их локализация определяет тип наследования болезни:
- X-сцепленный доминантный. Связан с мутацией в локусе COL4A5, который находится на половой хромосоме X. Ген кодирует а5-цепь коллагена 4 типа. Данный генетический дефект обуславливает 80-85% случаев наследственного нефрита. В полной мере заболевание проявляется у мальчиков и мужчин, у представительниц женского пола оставшийся нормальный ген в X-хромосоме компенсирует производство функционального коллагена.
- Аутосомно-рецессивный. Развивается на основе мутаций в генах C0L4A3 и COL4A4. Они локализованы на второй хромосоме, отвечают за структуру а3- и а4-цепи коллагена. Пациенты с этим вариантом синдрома составляют около 15% больных. Выраженность симптомов не зависит от пола.
- Аутосомно-доминантный. Нефрит возникает в результате мутаций генов COL4A3-COLA4, находящихся на 2 хромосоме. Как и в случае аутосомно-рецессивной формой болезни, нарушается синтез а4- и а3-цепей коллагена четвертого типа. Распространенность – 1% всех случаев генетического нефрита.
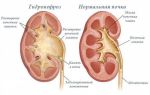
Гломерулярная базальная мембрана имеет сложное строение, ее образует строгая геометрическая последовательность молекул коллагена 4-го типа и полисахаридные компоненты. При синдроме Альпорта имеются мутации, которые задают дефектное строение спиралевидных коллагеновых молекул.
На первых этапах болезни базальная мембрана истончается, начинает расщепляться и расслаиваться. Одновременно возникают утолщенные участки с неравномерными просветлениями. Внутри скапливается тонкогранулярное вещество.
Прогрессирование болезни сопровождается полным разрушением базальной гломерулярной мембраны клубочковых капилляров, канальцев почек, структур внутреннего уха и глаз.
Таким образом, патогенетически синдром Альпорта представлен четырьмя звеньями: мутацией гена, дефектом строения коллагена, деструкцией базальных мембран, патологией почек (иногда – нарушением слуха и зрения).
Самым распространенным проявлением синдрома Альпорта является гематурия. Микроскопически этот симптом определяется у 95% женщин и у 100% мужчин. При рутинном обследовании мальчиков гематурия обнаруживается уже в первые годы жизни. Другой распространенный признак заболевания – протеинурия.
Выведение белка с мочой у пациентов мужского пола с X-сцепленным синдромом начинается в раннем детском возрасте, у остальных – позже. У девочек и женщин уровень экскреции белка повышается незначительно, случаи выраженной протеинурии крайне редки.
У всех больных отмечается неуклонное прогрессирование симптома.
Артериальная гипертензия характерна для мужчин с классическим типом синдрома и для пациентов обоих полов с аутосомно-рецессивным вариантом наследования. Тяжесть гипертонии увеличивается вместе с нарастанием ХПН.
У юношей, мужчин снижение функции почек достигает терминальной стадии к 16-35 годам, при медленном течении болезни – к 45-65 годам.
Иногда выявляются диффузные гладкомышечные опухоли пищевода и бронхов, проявляющиеся в позднем детстве дисфагией, рвотой, болями в эпигастрии и за грудиной, одышкой, частыми бронхитами.
Часто у больных формируется нейросенсорная тугоухость. Нарушения слуха дебютируют в детстве, но становятся заметными в подростничестве или молодости. У детей тугоухость распространяется только на звуки высокой частоты, обнаруживается в специально созданных условиях – при аудиометрии.
По мере взросления и прогрессирования синдрома нарушается слуховое восприятие средних и низких частот, в том числе человеческой речи. При X-связанном синдроме расстройство слуха к 25 годам имеется у 50% больных мужчин, к 40 годам – у 90%. Тяжесть тугоухости вариабельна, от изменений только в результатах аудиограммы до полной глухоты.
Патологии вестибулярного аппарата отсутствуют.
Расстройства зрения включают передний лентиконус – выпячивание центра хрусталика глаза вперед и ретинопатию. Обе патологии проявляются прогрессирующим ухудшением зрительной функции, покраснением, болью в глазах.
У некоторых больных имеются стигмы дизэмбриогенеза – анатомические аномалии мочевыделительной системы, глаз, ушных раковин, конечностей.
Может наблюдаться высокое расположение неба, укорочение и искривление мизинцев, сращивание пальцев ног, широко расставленные глаза.
Отсутствие лечения больных синдромом Альпорта приводит к быстрому прогрессированию глухоты и слепоты, формированию катаракт. У части пациентов развивается полиневропатия – поражение нервов, сопровождающееся мышечной слабостью, болями, судорогами, тремором, парестезиями, снижением чувствительности.
Другим осложнением является тромбоцитопения с высоким риском кровотечений. Наиболее опасным состоянием при наследственном нефрите считается терминальная стадия почечной недостаточности. Больше всего ей подвержены мужчины с типом наследования, сцепленным с половой X-хромосомой.
К 60 годам 100% больных этой группы нуждаются в процедурах гемодиализа, перитонеального диализа, трансплантации донорской почки.
В диагностическом процессе принимают участие врачи-нефрологи, урологи, терапевты и генетики. При опросе выясняется возраст дебюта симптомов, наличие у родственников первой линии гематурии, протеинурии или смертельных исходов вследствие ХПН.
Для синдрома Альпорта характерно раннее начало и отягощенный семейный анамнез. Дифференциальная диагностика направлена на исключение гематурической формы гломерулонефритов, вторичных нефропатий.
Для подтверждения диагноза проводятся следующие процедуры:
- Физикальное обследование. Определяется бледность кожных покров и слизистых оболочек, сниженный мышечный тонус, внешние и соматические признаки дизэмбриогенеза – высокое небо, аномалии строения конечностей, увеличенное расстояние между глазами, сосками. На ранних стадиях болезни диагностируется артериальная гипотония, на поздних – артериальная гипертония.
- Общий анализ мочи. Обнаруживаются эритроциты и повышенное содержание белка – признаки гематурии и протеинурии. Показатель белка мочи напрямую коррелирует с тяжестью синдрома, по его изменению оценивается прогрессирование патологии, вероятность нефротического синдрома, ХПН. Возможно наличие признаков лейкоцитурии абактериального характера.
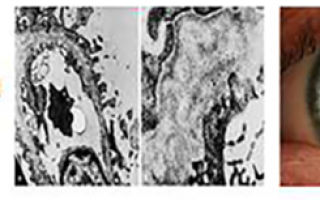
- Исследование биоптата почек. При микроскопии визуализируется истонченная базальная мембрана, расщепление и разделение ее слоев. На поздней стадии отмечаются утолщенные дистрофичные участки с «сотами» просветления, зоны полной деструкции слоя.
- Молекулярно-генетическое исследование. Генетическая диагностика не является обязательной, но позволяет составить более точный прогноз, подобрать оптимальную схему лечения. Изучается строение генов, мутации в которых обуславливают развитие синдрома. У большей части больных выявляются мутации гена COL4A5.
- Аудиометрия, офтальмологическое исследование. Дополнительно пациентам могут быть назначены диагностические консультации сурдолога и офтальмолога. При аудиометрии обнаруживается снижение слуха: в детском и подростковом возрасте – билатеральная высокочастотная тугоухость, во взрослом возрасте – низкочастотная и среднечастотная тугоухость. Офтальмолог определяет искажение формы хрусталика, поражение сетчатки, наличие катаракты, снижение зрения.
Специфическая терапия отсутствует. С раннего возраста проводится активное симптоматическое лечение, снижающее протеинурию. Оно позволяет предотвратить поражение и атрофию почечных канальцев, развитие интерстициального фиброза.
С помощью ингибиторов ангиотензинпревращающего фермента и блокаторов рецепторов к ангиотензину II удается приостановить прогрессирование заболевания, добиться регрессии гломерулосклероза, тубулоинтерстициальных и сосудистых изменений в почках.
Пациентам с терминальной стадией ХПН назначается гемодиализ, перитонеальный диализ, решается вопрос о целесообразности трансплантации почек.
Синдром прогностически благоприятен в случаях, когда гематурия протекает без протеинурии, нет расстройств зрения и тугоухости. Кроме этого, прогноз хороший у большинства женщин – даже при наличии гематурии болезнь прогрессирует медленно, не ухудшает общего состояния.
Ввиду наследственного характера патологии предупредить ее развитие невозможно. В семьях, где установлено наличие X-сцепленной формы синдрома, возможно проведение пренатальной диагностики. Генетический скрининг особенно рекомендован женщинам, вынашивающим мальчиков.
Источник: https://www.KrasotaiMedicina.ru/diseases/genetic/Alport-syndrome
Синдром Альпорта — причины и симптомы, диагностика и лечение синдрома Альпорта
27486

Синдром Альпорта (семейный гломерулонефрит) – это редкое генетическое заболевание, которое характеризуется гломерулонефритом, прогрессирующей почечной недостаточностью, нейросенсорной тугоухостью и поражением глаз. Заболевание было впервые описано британским врачом Артуром Альпортом в 1927 году. Синдром Альпорта встречается очень редко, но в США он отвечает за 3% случаев терминальной почечной недостаточности у детей и 0,2% у взрослых, а также считается наиболее распространенным типом семейного нефрита.
Тип наследования синдрома Альпорта может быть разным:
• Х-сцепленный доминантный (XLAS): 85%. • Аутосомно-рецессивный (ARAS): 15%. • Аутосомно-доминантный (ADAS): 1%. Наиболее распространенная Х-сцепленная форма синдрома Альпорта приводит к терминальной стадии почечной недостаточности у мужчин. Гематурия обычно возникает у мальчиков с синдромом Альпорта в первые годы жизни. Протеинурия обычно отсутствует в детстве, но это состояние часто развивается у мужчин с XLAS и у представителей обоих полов с ARAS. Потеря слуха и поражение глаз никогда не обнаруживаются при рождении – они возникают в позднем детстве или в юности, незадолго до развития почечной недостаточности. Синдром Альпорта вызван мутациями в генах COL4A4, COL4A3, COL4A5, отвечающих за биосинтез коллагена. Мутации в указанных генах нарушают нормальный синтез коллагена типа IV, который является очень важным структурным компонентом базальных мембран в почках, внутреннем ухе и глазах. Базальные мембраны – это тонкие пленочные структуры, которые поддерживают ткани и отделяют их друг от друга. При нарушении синтеза коллагена типа IV гломерулярные базальные мембраны в почках не способны нормально фильтровать токсичные продукты из крови, пропуская в мочу белки (протеинурия) и эритроциты (гематурия). Аномалии синтеза коллагена типа IV приводят к почечной недостаточности и отказу почек, что и является главной причиной смерти при синдроме Альпорта. Гематурия – это наиболее частое и раннее проявление синдрома Альпорта. Микроскопическая гематурия наблюдается у 95% женщин и практически у всех мужчин. У мальчиков гематурию обычно выявляют в первые годы жизни. Если у мальчика за первые 10 лет жизни не обнаружена гематурия, то американские эксперты рекомендуют считать, что у него маловероятно наличие синдрома Альпорта. Протеинурия в детстве обычно отсутствует, но иногда развивается у мальчиков с Х-сцепленным синдромом Альпорта. Протеинурия, как правило, прогрессирует. Значительная протеинурия у больных женского пола встречается нечасто. Гипертензия чаще присутствует у пациентов мужского пола с XLAS и у больных обоих полов с ARAS. Частота и тяжесть гипертензии повышается с возрастом и по мере прогрессирования почечной недостаточности. Нейросенсорная тугоухость (нарушение слуха) – это характерное проявление синдрома Альпорта, которое наблюдается довольно часто, но не всегда. Есть целые семьи с синдромом Альпорта, которые страдают от тяжелой нефропатии, но имеют нормальный слух. Нарушение слуха никогда не обнаруживается при рождении. Билатеральная высокочастотная нейросенсорная тугоухость обычно проявляется в первые годы жизни или в раннем подростковом возрасте. На ранней стадии болезни нарушение слуха определяется только при аудиометрии. По мере прогрессирования, нарушение слуха распространяется на низкие частоты, включая человеческую речь. После появления тугоухости следует ожидать вовлечения почек. Американские ученые утверждают, что при Х-связанном синдроме Альпорта 50% мужчин страдают нейросенсорной тугоухостью к 25 годам, а к 40 годам – около 90%. Передний лентиконус (выпячивание центрального участка хрусталика глаза вперед) наблюдается у 25% пациентов с XLAS. Лентиконуса нет при рождении, но с годами он приводит к прогрессирующему ухудшению зрения, которое заставляет больных часто менять очки. Состояние не сопровождается болью в глазах, покраснением или нарушениями цветового зрения. Ретинопатия – это самое распространенное проявление синдрома Альпорта со стороны органа зрения, поражает 85% мужчин с Х-сцепленной формой болезни. Появление ретинопатии обычно предшествует почечной недостаточности. Задняя полиморфная дистрофия роговицы – редкое состояние при синдроме Альпорта. У большинства нет никаких жалоб. Мутация L1649R в гене коллагена COL4A5 может также вызывать истончение сетчатки, которое ассоциируется с Х-сцепленным синдромом Альпорта. Диффузный лейомиоматоз пищевода и бронхиального дерева – еще одно редкое состояние, которое наблюдается в некоторых семьях с синдромом Альпорта. Симптомы появляются в позднем детском возрасте и включают нарушение глотания (дисфагия), рвоту, боль в эпигастрии и за грудиной, частые бронхиты, одышку, кашель. Лейомиоматоз подтверждается компьютерной томографией или МРТ. На ARAS приходится всего 10-15% случаев болезни. Эта форма встречается у детей, чьи родители являются носителями одного из пораженных генов, сочетание которых у ребенка вызывает болезнь. Сами родители не имеют симптомов или имеют незначительные проявления, а дети тяжело больны – их симптомы напоминают XLAS. ADAS – это самая редкая форма синдрома, которая затрагивает одно поколение за другим, причем мужчины и женщины болеют одинаково тяжело. Почечные проявления и глухота напоминают XLAS, но почечная недостаточность может возникать в более позднем возрасте. Клинические проявления ADAS дополняются склонностью к кровотечениям, макротромбоцитопенией, синдромом Эпштейна, наличием нейтрофильных включений в крови. • Лабораторные анализы. Анализ мочи: у больных с синдромом Альпорта чаще всего присутствует кровь в моче (гематурия), а также высокое содержание белка (протеинурия). Анализы крови демонстрирует почечную недостаточность. • Биопсия тканей. Ткань почек, полученную при биопсии, исследуют с помощью электронной микроскопии на наличие ультраструктурных аномалий. Биопсия кожи менее инвазивна, и американские эксперты рекомендуют выполнять ее в первую очередь. • Генетический анализ. В диагностике синдрома Альпорта, если остаются сомнения после биопсии почки, генетический анализ используется для получения однозначного ответа. Определяются мутации генов синтеза коллагена типа IV. • Аудиометрия. Все дети с семейной историей, позволяющей заподозрить синдром Альпорта, должны проходить высокочастотную аудиометрию для подтверждения нейросенсорной тугоухости. Рекомендуется периодический мониторинг. • Обследование глаз. Обследование у офтальмолога очень важно для раннего выявления и мониторинга переднего лентиконуса и других аномалий. • УЗИ почек. На поздних стадиях синдрома Альпорта ультразвуковое исследование почек помогает выявить структурные нарушения. Британские специалисты, основываясь на новых данных (2011) по генетическим мутациям у пациентов с Х-сцепленным синдромом Альпорта, рекомендуют анализ на мутации гена COL4A5, если пациент отвечает хотя бы двум диагностическим критериям по Gregory, и анализ COL4A3 и COL4A4, если мутация COL4A5 не обнаружена или подозревается аутосомное наследование. Синдром Альпорта пока неизлечим. Исследования показали, что ингибиторы АПФ могут уменьшать протеинурию и замедлять прогрессирование почечной недостаточности. Таким образом, использование ИАПФ целесообразно у пациентов с протеинурией, независимо от наличия гипертензии. То же самое касается антагонистов АТII-рецепторов. Оба класса препаратов, судя по всему, помогают уменьшить протеинурию путем снижения внутриклубочкового давления. Более того, ингибирование ангиотензина-II, ростового фактора, отвечающего за гломерулярный склероз, теоретически может замедлять склерозирование. Некоторые исследователи предполагают, что циклоспорин способен уменьшать протеинурию и стабилизировать почечную функцию у пациентов с синдромом Альпорта (исследования были небольшими). Но отчеты говорят, что ответ пациентов на циклоспорин очень вариабельный, и иногда препарат может ускорять интерстициальный фиброз. При почечной недостаточности стандартная терапия включает эритропоэтин для лечения хронической анемии, препараты для контроля остеодистрофии, коррекцию ацидоза и антигипертензивную терапию для контроля АД. Применяется гемодиализ и перитонеальный диализ. Больным с синдромом Альпорта трансплантация почки не противопоказана: опыт пересадки в США показал хорошие результаты. Генная терапия при разных формах синдрома Альпорта является перспективным вариантом лечения, который сегодня активно изучается западными медицинскими лабораториями.
Константин Моканов: магистр фармации и профессиональный медицинский переводчик
- Новости из медицинских лабораторий Европы и США — сентябрь 2013 Химический компонент, обычно используемый в качестве консерванта для собачьего корма, способен предотвращать развитие периферической нейропатии, связанной с приемом паклитаксела (Taxol). Медицина
- Пневмококковые инфекции — эпидемиология и пути заражения, лечение и профилактика Всемирная Организация Здравоохранения (ВОЗ) разместила пневмококковую инфекцию рядом с малярией на первом месте в списке инфекционных заболеваний, которым необходимо уделить пристальное внимание. Тяжелая пневмококковая инфекция грозит пожилым и хронически больным людям, а также детям. Медицина
 Новости из медицинских лабораторий Европы и США — сентябрь 2013 Химический компонент, обычно используемый в качестве консерванта для собачьего корма, способен предотвращать развитие периферической нейропатии, связанной с приемом паклитаксела (Taxol). Медицина
Новости из медицинских лабораторий Европы и США — сентябрь 2013 Химический компонент, обычно используемый в качестве консерванта для собачьего корма, способен предотвращать развитие периферической нейропатии, связанной с приемом паклитаксела (Taxol). Медицина  Пневмококковые инфекции — эпидемиология и пути заражения, лечение и профилактика Всемирная Организация Здравоохранения (ВОЗ) разместила пневмококковую инфекцию рядом с малярией на первом месте в списке инфекционных заболеваний, которым необходимо уделить пристальное внимание. Тяжелая пневмококковая инфекция грозит пожилым и хронически больным людям, а также детям. Медицина
Пневмококковые инфекции — эпидемиология и пути заражения, лечение и профилактика Всемирная Организация Здравоохранения (ВОЗ) разместила пневмококковую инфекцию рядом с малярией на первом месте в списке инфекционных заболеваний, которым необходимо уделить пристальное внимание. Тяжелая пневмококковая инфекция грозит пожилым и хронически больным людям, а также детям. Медицина Источник: https://medbe.ru/news/meditsina/sindrom-alporta-prichiny-i-simptomy-diagnostika-i-lechenie-sindroma-alporta/
Синдром Альпорта: причины, симптомы, классификация, лечение, прогнозы
Наследственный нефрит (более известное название – синдром Альпорта) – патология достаточно редкая.
По официальным данным, в России на 100 000 новорожденных малышей приходится 17 с такой аномалией развития.
В Европе 1% всех больных хронической почечной недостаточностью (ХПН) – это именно люди с наследственным нефритом. А 2,3% операций по пересадке почки делают пациентам с этим диагнозом.
Синдром Альпорта?
Наследственный нефрит – это постоянно прогрессирующее заболевание почек, которое часто идет параллельно с потерей слуха и серьезными проблемами со зрением. В справочниках можно найти определение синдрома Альпорта (СА) как неиммунной наследственной формы гломерулопатии, то есть поражения клубочкового аппарата почек.
Врожденное расстройство почечной функции проявляется у детей уже в 3-5 лет, возникает из-за мутаций одного их трех генов, которые отвечают за выработку коллагена IV типа.
Четвертая коллагеновая разновидность составляет основу базальных мембран почечных клубочков, кохлеарного аппарата (часть внутреннего уха), а также капсулы хрусталика. Отсюда – и одновременные нарушения работы почек, слуха и зрения.
Международная классификация болезней 10-го пересмотра (МКБ-10) – главный нормативный документ, систематизирующий все существующие нарушения здоровья, – относит детское заболевание к классу врожденных аномалий, деформаций и хромосомных нарушений.
Поскольку при СА страдает целый ряд органов, то болезнь входит в группу врожденных пороков, затрагивающих сразу несколько систем. И маркируется кодом Q87.8 – это «другие уточненные синдромы врожденных аномалий, не классифицированные в других рубриках».
Причины
Чаще всего поврежденный ген передается малышу от родителей. Когда болезнь почек переходит по Х-хромосоме, то мать может стать передатчиком аномалии и сыну, и дочери. Отец – только дочери. Вероятность того, что малыш родится с поражением почек, увеличивается в разы, если в роду есть люди с болезнями мочевыделительной системы (в первую очередь ХПН).
Но в 20% случаев дети с синдромом Альпорта рождаются в семьях, где у всех родственников идеально здоровые почки. Здесь речь идет о случайных, спонтанных генетических мутациях.
Симптомы
Врожденный наследственный нефрит развивается при недостатке коллагена, одного из главнейших структурных элементов соединительной ткани. В результате коллагенового дефицита базальные мембраны почечных клубочков, внутреннего уха и глазного аппарата истончаются и расщепляются, а сами органы перестают полноценно справляться со своей функцией.
Все симптомы синдрома Альпорта распределяются на два группы – почечные и внепочечные проявления. В числе почечных диагностируются два главных признака: гематурия (следы крови в моче) и протеинурия (белки в моче). Нередко их объединяют под названием «изолированный мочевой синдром».
Изолированный мочевой синдром у ребятишек можно заметить не сразу. Видимые сигналы появляются только на 3-5-м году жизни, бывает и в 7-10 лет. Но мельчайшие капельки крови в моче присутствуют всегда, даже если поначалу их не видно – это бессимптомная микрогематурия. Поэтому гематурия считается основным специфическим признаком синдрома Альпорта.
Примерно в половине случаев спусковым крючковым для появления крови в детской моче служит инфекция. Почечные симптомы появляются через 1-2 дня после ОРВИ. У мальчиков также развивается протеинурия, но позже, обычно после 10 лет. У девочек этот симптом или сглажен, или его вовсе нет.
Внепочечные симптомы врожденного нефрита проявляются позднее. Это:
- тугоухость (сначала ребенок перестает различать высокие звуки, потом обычную речь);
- различные глазные нарушения;
- отставание в физическом развитии;
- врожденные аномалии (деформированные уши, высокое небо, сращенные или дополнительные пальчики – не более 7 признаков);
- редко – лейомиоматоз (разрастание гладких волокон мышц) пищевода, трахеи, бронхов.
По мере прогрессирования недуга появляются классические признаки почечной недостаточности: желтоватая и сухая кожа, сухость во рту, уменьшение количества мочи и т.д. Увеличивается давление в почечных сосудах – гипертензия.
Классификация
Существует две классификации синдрома Альпорта у детей. Первая – генетическая, по типу наследования аномалии.
В соответствии с этой классификацией выделяют три типа врожденного нефрита:
- Х-сцепленный доминантный, или классический (около 80% всех пациентов с СА);
- аутосомно-рецессивный (15% детей с врожденной аномалией);
- аутосомно-доминантный (самый редкий тип, около 5% пациентов).
Вторая, основная классификация называет три варианта почечного заболевания:
- Нефрит, сопровождающийся гематурией, тугоухостью и проблемами со зрением (поражения глаз). Это Х-доминантный тип врожденного порока.
- Нефрит с гематурией, но без поражения органов чувств. Соответствует аутосомно-рецессивной форме.
- Доброкачественная семейная гематурия.
Первые два варианта – это прогрессирующее почечное заболевание, неизбежный итог которого – хроническая почечная недостаточность. При доброкачественной семейной гематурии ХПН не развивается, а качество и продолжительность жизни никак не страдают.
Диагностика
К таким признакам относят:
- В семье есть случаи гематурии, в роду встречались случаи летального исхода от хронической почечной недостаточности.
- В семье у ребенка диагностирована гематурия и/или протеинурия.
- Специфические изменения у пациента базальной мембраны почечных клубочков (по результатам биопсии).
- Врожденная патология зрения.
- Падение слуха (выявляется по данным аудиометрии).
При подозрении на синдром Альпорта используется ряд традиционных диагностических методов:
- сбор анамнеза (сведения о наличии тех же симптомов и смертей от ХПН у кровных родственников);
- физикальные способы (пальпация, простукивание);
- лабораторные анализы (клинический анализ мочи и др.);
- УЗИ и биопсия почки.
Специалисты также рекомендуют ДНК-тест для членов семьи юного пациента с использованием ДНК-зондов. Это позволяет определить носителя мутантного гена. Кроме того, существует возможность применения ДНК-зондов и для предродовой диагностики синдрома Альпорта, еще во время беременности мамы. Это особенно важно, если в семье ждут мальчика, – у мужчин СА протекает тяжелее.
В обязательном порядке требуется и дифференциальная диагностика: для отграничения врожденного нефрита от нефропатии и приобретенного гломерулонефрита.
Лечение
На начальном этапе врожденного нефрита мощная комплексная терапия не требуется.
При постановке почечного диагноза необходимы следующие терапевтические меры для детей:
- отсутствие серьезных физических нагрузок (освобождение от уроков физкультуры);
- постоянные прогулки;
- сбалансированное питание;
- фитотерапия при появлении крови в детской моче (настой крапивы и тысячелистника, сок черноплодной рябины);
- витамины А и Е, В6 (пиридоксин) для улучшения обмена веществ (курсами по 2 недели);
- для этих же целей – инъекции кокарбоксилазы.
Мальчикам для снижения протеиноурии рекомендуются ингибиторы АТФ (ангиотензин-превращающего фермента) и блокаторы рецепторов ангиотензина.
Когда хроническая почечная недостаточность переходит в самую опасную, терминальную, стадию, требуется постоянный гемодиализ. В самых тяжелых случаях – пересадка почки.
Прогноз при синдроме Альпорта зависит от двух факторов: варианта болезни и пола ребенка. Быстрее всего прогрессирует классическая, Х-доминантная форма синдрома Альпорта у мальчиков.
В этом случае ХПН диагностируют у всех больных до 60 лет, а у 50% — до 25 лет. Если в семье есть мужчины с таким же вариантом нефрита, то время наступления терминальной стадии почечной недостаточности можно легко спрогнозировать, оно будет таким же. У женщин такой зависимости нет.
При аутосомно-рецессивном типе почечная недостаточность развивается немного медленнее, но есть риск того, что ХПН перейдет в терминальную стадию уже к 30 годам.
При аутосомно-доминантной форме течение и прогнозы наиболее благоприятные: хронической почечной недостаточности ситуация не доходит. Эта форма соответствует доброкачественной семейной гематурии. Специфическая терапия в этом случае не проводится, наличие крови в моче не угрожает жизни человека. Необходим лишь постоянный врачебный контроль за состоянием пациента.
Источник: http://gidmed.com/nefrologiya/zabolevaniya-nefr/nefrity/sindrom-alporta.html
Синдром Альпорта
Синдром Альпорта — это наследственное заболевание, характеризующееся прогрессирующем снижением функции почек в сочетании с патологией слуха и зрения. В России частота распространения заболевания среди детского населения составляет 17:100000.
Причины синдрома Альпорта
Установлено, что за развитие заболевания ответственен ген, который находится в длинном плече Х хромосомы в зоне 21-22 q. Причиной болезни является нарушение структуры коллагена IV типа.
Коллаген -это белок, основной компонент соединительной ткани, который обеспечивает её прочность и эластичность.
В почках выявляется дефект коллагена сосудистой стенки, в области внутреннего уха -кортиева органа, глаза — капсулы хрусталика.
Симптомы синдрома Альпорта
При синдроме Альпорта отмечается значительная вариабельность внешних проявлений. Как правило, заболевание манифестирует в возрасте 5-10 лет с гематурии (появление крови в моче). Обычно гематурия выявляется случайно при обследовании ребенка.
Гематурия может протекать с наличием или отсутствием протеинурии (появление белка в моче). При выраженной потере белка может развиваться нефротический синдром, который характеризуется отеками, повышением артериального давления, симптомами отравления организма вредными продуктами при снижении функции почек.
Возможно повышение количества лейкоцитов в моче при отсутствии бактерий.
У большинства больных обращают на себя внимание стигмы дизэмбриогенеза. Стигмы дизэмбриогенеза — это небольшие внешние отклонения, которые существенно не сказываются на функционировании организма. К ним относятся: эпикант (складка у внутреннего угла глаза), деформация ушных раковин, высокое небо, увеличение количества пальцев или их сращение.
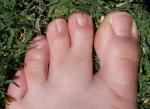
Эпикант. Синдактилия.
Очень часто одинаковые стигмы дисэмбриогенеза выявляются у заболевших членов семьи.
Снижение слуха в результате неврита слухового нерва также характерно для синдрома Альпорта. Тугоухость чаще развивается у мальчиков и иногда выявляется раньше, чем поражение почек.
Аномалии зрения проявляются в виде лентиконуса (изменение формы хрусталика), сферофакии (шаровидная форма хрусталика) и катаракты (помутнение роговицы).
Симптомы заболевания почек выявляются, как правило, в подростковом возрасте. Хроническая почечная недостаточность диагностируется в совершеннолетнем возрасте. Иногда возможно быстрое прогрессирование заболевания с формированием уже в детском возрасте терминальной почечной недостаточности.
Диагностика синдрома Альпорта
- Предположить синдром Альпорта можно на основании данных родословной по наличию заболевания у других членов семьи. Для диагностики заболевание необходимо выявление трех из пяти критериев:
- • наличие гематурии или смертность от хронической почечной недостаточности в семье;• наличие гематурии и/или протеинурии у членов семьи;• выявление специфических изменений при биопсии почек;• тугоухость;
- • врожденная патология зрения.
- В настоящее время также рекомендуется проведение ДНК-диагностики с целью выявления дефектного гена.
Лечение синдрома Альпорта
В условиях отсутствия специфического лечения, главной целью становится замедление развития почечной недостаточности. Детям запрещаются физические нагрузки, назначается полноценное сбалансированное питание.
Особое внимание уделяется санации инфекционных очагов. Применение гормональных препаратов и цитостатиков не приводит к значимому улучшению состояния. Основным методом лечения остается трансплантация (пересадка) почки.
- Неблагоприятный прогноз течения заболевания, который характеризуется быстрым развитием терминальной почечной недостаточности, наиболее вероятен при наличии следующих критериев:
- — мужской пол;- высокая концентрация белка в моче;- раннее развитие нарушений функции почек у членов семьи;
- — тугоухость.
- При выявлении изолированной гематурии без протеинурии и нарушения слуха прогноз течения заболевания благоприятный, почечная недостаточность не формируется.
Врач терапевт, нефролог Сироткина Е.В.
Источник: https://medicalj.ru/diseases/congenital-anomaly/934-sindrom-alporta
Синдром Альпорта: что это, симптомы и лечение
Синдром Альпорта – наследственное поражение почек прогрессирующего характера. В патогенезе заболевания лежит дефект коллагеновых волокон. Проявления варьируются в зависимости от формы хромосомного заболевания и гендерной принадлежности больного.
Общие сведения и этиопатогенез
Синдром Альпорта может развиваться по двум механизмам:
- Мутация Х-хромосомы (85% случаев)
- Аутосомная мутация (15% случаев)
Наследственная нефропатия является одним из самых частых хромосомных заболеваний человечества. Аномальная генетическая информация может передаваться от родителей к ребенку, или возникать спонтанно у ребенка полностью здоровой семьи.
Поврежденная Х-хромосома имеет разное влияние на проявления заболевания в зависимости от пола ребенка. По причине того, что у женщин есть не одна, а две Х-хромосомы, нормальная хромосома нивелирует действие патологической, что значительно уменьшает проявления.
У мужчин есть одна Х-хромосома и одна У-хромосома. Поэтому у мужского пола проявления синдрома значительно ярче выражены и несут более тяжелые последствия.
Аутосомная мутация возникает значительно реже. Поврежденный генетический материал приводит к развитию ярко выраженных симптомов вне зависимости от пола. К счастью, данная форма синдрома Альпорта возникает значительно реже, и лишь в отдельных случаях передаётся наследственно.
Повреждение аутосомы или гетеросомы приводит к нарушению экспрессии гена. Вследствие этого синтезируется дефектный коллаген четвертого типа. Коллаген IV очень важен для организма человека. Он является составной частью клеточных мембран, входит в состав хрусталика глаза и каналов внутреннего уха.
Клиническая картина
Проявления синдрома Альпорта отличаются между собой выраженностью проявлений. В большинстве случаев на передний план выступает прогрессирующая нефропатия.
Больной может жаловаться на длительную давящую боль в пояснице. Болевой синдром постоянный, может иррадиировать в область паха или ногу. Использование обезболивающих препаратов снижает интенсивность болевых ощущений, но полностью их не снимает.
Эпизоды болевого синдрома могут обостряться после переохлаждения, значительных физических нагрузок.
- Отеки лица утром появляются при прогрессировании болезни. Лицо становится пастозным, вокруг глаз появляться мешки. Во второй половине дня отечность уменьшается, иногда полностью исчезает.
- Увеличенное количество мочи. Зачастую больные с нефропатией отмечают, что количество выделяемой мочи даже больше количества выпитой жидкости.
- Нарушение зрения. Возникает у половины больных синдромом Альпорта. Может проявляться катарактой в молодом возрасте, вывихом хрусталика глаза.
- Нарушение слуха. В 25-30% случаев возникает сенсорная глухота, которая имеет тенденцию к прогрессированию. Чаще процесс двусторонний и симметрический. Лишь в редких случаях приводит к полной потере слуха.
Постановка диагноза
Во время общения врача и пациента нужно отвести особое внимание семейному анамнезу. Важно выяснить, были ли подобные симптомы у близких родственников, как они проявлялись, и поддавались ли лечению.
Для подтверждения диагноза проводят лабораторные и инструментальные методы исследования. В общем анализе мочи отмечается появление эритроцитов, лейкоцитов, белка.
Причина их появления заключается в нарушении барьерной функции нефронов, которые не способны удерживать большие элементы крови в сосудах. Биохимический анализ крови показывает гипогаммаглобулинемию, общую гипопротеинемию.
Самый точный, обязательный, метод исследования при подозрении на синдром Альпорта – биопсия с последующим кариотипированием хромосом. Материал чаще берут прицельной биопсией пораженной почки. При аутосомном типе заболевания биопсический материал можно взять и со слизистых оболочек ротовой полости или глотки.
Синдром Альпорта у детей
Наследственная нефропатия в детском возрасте возникает очень редко. Практически во всех случаях совмещается с другими хромосомными аномалиями – синдромом Дауна, синдромом Клайнфельтера и так далее.
Клинические проявления и лабораторная диагностика полностью схожи с ситуацией у взрослых, но есть одно важное отличие. У взрослых болезнь начинает свои проявления в возрасте 20-30 лет, тогда как у ребенка первые симптомы могут появиться уже в возрасте 2-х лет. Лечение аналогично взрослой форме и направленно на предотвращение прогрессирования осложнений.
Последствия и осложнения синдрома Альпорта
У мужчин болезнь несет более тяжелый характер, намного быстрее приводит к осложнениям.
Самые частые осложнения:
- Нефротический синдром.
- Хроническая почечная недостаточность.
- Снижение или потеря зрения.
- Снижение или потеря слуха.
Все вышеперечисленные синдромы не обязательно должны проявиться у одного человека, могут совмещаться в разных комбинациях и отличаться по выраженности проявлений.
Женщины при наличии аномалии Х-хромосомы компенсируют генетическую аномалию другой, здоровой Х-хромосомой, поэтому проявления и осложнения болезни у них бывают крайне редко.
Отличить здоровую женщину от женщины с патологией можно лишь лабораторно. В анализе мочи появляются эритроциты и следы белка. Методом кариотипирования можно подтвердить диагноз.
При более редкой, аутосомной форме, симптомы заболевания у женщин будут возникать аналогично мужчинам. Осложнения в таком случае появляются уже в возрасте 20-25 лет, несут опасный для жизни характер.
Методы лечения и профилактики
Современная медицина не способна изменить первопричину заболевания – хромосомную аномалию. Поэтому терапевтические методы направленны на устранение последствий.
Хроническая почечная недостаточность поддается лечению концентратами альбуминов. Для уменьшения прогрессии показана диета, богатая на животные и растительные белки (мясо, рыба, картофель, свекла, капуста, морковь). Нужно минимизировать прием соли до 2-х грамм в сутки.
Медикаментозное лечение:
- Глюкокортикостероиды (Преднизолон, Бетаметазон, Флуметазон )
- Диуретики (Фуросемид)
- Антикоагулянты.
- Ингибиторы АПФ (Рамиприл, Еналаприл)
Глазные симптомы чаще лечатся оперативно – замена хрусталика глаза. Глухота, к сожалению, не поддается медикаментозной и хирургической терапиям. Для улучшения слуха проводят постановку слухового аппарата.
С целью профилактики у семьи со случаями синдрома Альпорта стоит пройти консультацию генетика перед зачатием ребенка. Благодаря генетическим анализам генетик подтвердит или исключит возможность рождения ребенка с нефропатическим синдромом и предоставит медицинские рекомендации.
Загрузка…
Источник: https://KardioBit.ru/pochki/lechenie/sindrom-alporta
Синдром Альпорта — диагностика и лечение. Симптомы и прогноз
Синдром Альпорта — это вид генетической патологии, проявляющейся недостаточным количеством синтеза коллагена IV, который входит в состав почечных мембран, глазного хрусталика и органа слуха. Вследствие поражения наблюдаются патологические изменения со стороны данных органов.
Заболевание возникает в результате мутаций в составе гена, кодирующего выработку коллагеновых волокон. Выделяют различные формы генных дефектов:
- делеция — выпадение одного из участков хромосомы;
- сплайсинг — нарушение образования активной мрнк, которая берет участие в переносе генетической информации;
- миссенс — точечная мутация, при которой триплет днк, которому соответствует определенная аминокислота, начинает кодировать другую аминокислотную цепь. Структурные нарушения ведут к функциональным изменениям;
- нонсенс — в состав цепи днк включается стоп-кодон там, где его в норме не должно быть, это приводит к остановке синтеза некоторых белков.
Распространенность синдрома Альпорта ничтожно мала — болеет один человек на пять тысяч населения.
Из-за вышеперечисленных процессов возникают структурные изменения в оболочках, которые в норме содержат в себе фрагменты коллагена, диагностируется их дисфункция. С прогрессированием болезни происходит истончение и расслаивание слоев оболочки, деструктивные изменения.
Симптомы синдрома Альпорта
Клинические признаки развиваются постепенно и выражаются следующим образом:
- стойкое повышение давление;
- выделение с мочой кровяных сгустков и протеиновых фракций;
- появление отеков;
- к глазным проявлениям симптома альпорта относят деформацию хрусталика, патологические изменения сетчатки;
- снижение остроты слуха начинается с плохого восприятия звуков высокой частоты, со временем человек не слышит не только посторонних звуков, но и речи собеседника;
- патологические изменения сетчатки;
- деформация хрусталика;
- ухудшение зрения;
- боль в глазах;
- покраснение конъюнктивы.
Классификация и виды синдрома Альпорта
Передача недуга происходит следующим образом:
- Х-сцепленный доминантный — в большинстве случаев недуг передается именно так. Это значит, что патология почек симптоматически проявляется только у представителей мужского пола так, как у них одна Х-хромосома. Женщины являются лишь носителями мутантного гена, вторая половая хромосома берет на себя все утраченные функции;
- аутосомно-рецесивный — с одинаковой вероятностью могут быть клинические проявления как у мужчин, так и у женщин. Как правило, диагностируют не у всех потомков, а через одно поколение;
- аутосомно-доминантный — проявляется в каждом семейном поколении, независимо от половой принадлежности, данный тип передачи встречается редко.
Диагностика синдрома Альпорта
Важным аспектом на этапе постановки диагноза является выяснение основных жалоб пациента. Одним из основных критерием отягощенная наследственность, то есть наличие данного заболевания у близких родственников.
Узкий специалист оценивает внешний вид больного —побледнение кожи, снижение мышечной силы, деформации костных структур лицевого черепа, кистей.
Среди анализов при синдроме Альпорта основным является изучение показателей мочи — здесь будет отмечаться протеинурия и выделение эритроцитов, цилиндров.
Чтобы оценить тип передачи, наличие заболевания у родственников, проводят генетико-молекулярные обследования: составление генеалогической карты, молекулярно-биохимические, цитологические методы.
Также проводится микроскопическое обследование взятых при биопсии почек тканевых фрагментов, чтобы изучить морфологию при синдрома Альпорта, — отмечается тонкость шаров оболочки почечных канальцев, зоны ее разрушения. Для выявления сопутствующих патологий органов чувств проводят аудиометрию, офтальмоскопию.
Лечение синдрома Альпорта
Этиологическая терапия отсутствует. Лечение направлено на снятие основной симптоматики и предупреждение дальнейшего прогрессирования болезни.
Применяются медикаменты для снижения уровня выделяющегося с мочой белка, а также препараты, которые предотвращают превращение ангиотезина I в II его форму и блокируют активность ангиотензина II.
Таким образом, останавливаются патологические процессы, приводящие к склерозирующим изменениям, структурным нарушениям и расстройству кровоснабжения почек.
Если заболевание выявлено на последних стадиях, рекомендуют применять гемодиализ. Оптимальным решением будет пересадка здорового органа.
Осложнения и прогноз синдрома Альпорта
Опасным последствием заболевания является развитие хронической почечной недостаточности. На начальных стадиях недуга, который появляется гематурией без выделения белковых фракций, если не наблюдаются нарушения слуховых и зрительных функций, прогноз благоприятный. При возникновении тяжелых дисфункциональных нарушений может быть назначена инвалидность при синдроме Альпорта.
Специфической профилактики недуга нет. Семьям, в которых предыдущие дети родились с такой патологией или в анамнезе близких родственников есть упоминание о ней, рекомендуют при планировании ребенка и во время беременности пройти комплексное молекулярно-генетическое обследование, чтобы оценить генетику и наследование синдрома Альпорта.
Источник: https://xmedicin.com/sindrom-alporta/
Синдром Альпорта: причины заболевания, основные симптомы, лечение и профилактика
Генетическая болезнь почек, при которой нарушается синтез коллагена 4 типа, отвечающего за образование базальных мембран нефронов, хрусталика и внутреннего уха. У мужчин наблюдаются более выраженные симптомы недуга. Как правило, у пациентов женского пола признаки патологии отсутствуют.
Заболевание проявляется микрогематурией, наличием белка в анализе мочи, почечной недостаточностью, сенсорной тугоухостью, катарактой, деформацией и вывихом хрусталика.
Чтобы установить и подтвердить диагноз, врач собирает анамнестические данные, анализирует клинические проявления, проводит физикальный осмотр и направляет больного на дополнительные обследования.
В рамках диагностики могут выполнять общее исследование мочи, биопсию почек с последующим микроскопическим исследованием, молекулярно-генетический анализ, аудиометрию и офтальмологический осмотр. Больному назначают ингибиторы ангиотензинпревращающего фермента и блокаторы рецепторов к ангиотензину.
Терминальная стадия хронической почечной недостаточности является показанием к гемодиализу и перитонеальному диализу. Возможна трансплантация почек. Если пациент не страдает от тугоухости и зрительных нарушений, а при гематурии отсутствует белок в моче, прогноз благоприятный.
Заболевание возникает на фоне дефектов генов COL4A5, C0L4A3, COL4A4, отвечающих за кодирование структуры коллагена 4 типа. Среди изменений генов выделяют миссенс, делеции, сплайсинг, нонсенс-мутации. Патология наследуется по Х-сцепленному доминантному, аутосомно-рецессивному и аутосомно-доминантному типу.
Симптомы синдрома Альпорта
Основным симптомом болезни считают гематурию. Обычно, кровь в моче выявляют во время рутинного обследования уже в раннем детском возрасте. Также патология проявляется выведением белка с мочой. Как правило, у пациентов женского пола наблюдается незначительное повышение белка в моче.
Выраженная протеинурия встречается редко. Со временем симптомы прогрессируют. Классическая форма синдрома и патология, наследуемая по аутосомно-рецессивному типу, проявляются артериальной гипертензии. По мере нарастания хронической почечной недостаточности гипертония становится более выраженной.
Терминальная стадия ХПН формируется до 35-ти лет, однако если недуг имеет медленное течение – после 45 лет. При диффузных гладкомышечных опухолях в бронхах и пищеводе отмечаются: дисфагия, рвота, боль в груди, одышка, частые бронхиты, эпигастральные боли.
Недуг может сопровождаться нейросенсорной тугоухостью, которая проявляется в детском возрасте и со временем нарастает. Кроме того, клиническая картина может дополняться передним лентиконусом, ретинопатией, стигмами дизэмбриогенеза, укорочением и искривлением мизинцев, сращиванием пальцев на ногах.
Заболевание может осложняться глухотой, слепотой, катарактой, полиневропатией, судорогами, тромбоцитопенией.
Диагностика синдрома Альпорта
Пациента консультируют специалисты нефрологического, урологического, терапевтического и генетического профилей.
Чтобы установить и подтвердить диагноз, врач собирает анамнестические данные, анализирует клинические проявления, проводит физикальный осмотр и направляет больного на дополнительные обследования.
В рамках диагностики могут выполнять общее исследование мочи, биопсию почек с последующим микроскопическим исследованием, молекулярно-генетический анализ, аудиометрию и офтальмологический осмотр. Кроме того, врачи исключают гематурическую форму гломерулонефрита и вторичные нефропатии.
Лечение синдрома Альпорта
Специфическое лечение не разработано. Больному назначают ингибиторы ангиотензинпревращающего фермента и блокаторы рецепторов к ангиотензину второго типа.
С помощью терапии гломерулосклероз, тубулоинтерстициальные и сосудистые изменения в почках регрессируют.
Терминальная стадия хронической почечной недостаточности является показанием к гемодиализу и перитонеальному диализу. Возможна трансплантация почек.
Профилактика синдрома Альпорта
Специальных превентивных мер не существует. Парам, планирующим зачатие, рекомендована консультация генетика. Если в семейном анамнезе выявлена Х-сцепленная форма синдрома, необходима пренатальная диагностика.
Источник: https://www.obozrevatel.com/health/bolezni/sindrom-alporta.htm