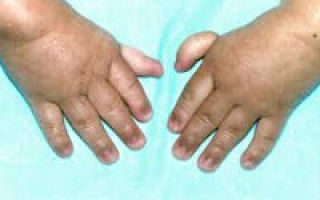
Синдром Фанкони – врожденная патология обмена веществ, которая заключается в прекращении всасывания почечными канальцами (реабсорбции) глюкозы, солей фосфорной и угольной кислоты, аминокислот. В результате формируется патология, которую можно отнести к особенному виду диабета или рахиту.
Другое более подробное наименование болезни – синдром де Тони-Дебре-Фанкони или глюкозо-фосфат-аминовый диабет – связано с уточнением описания заболевания тремя авторами. Поскольку первым был все-таки педиатр из Швейцарии Фанкони, который обнаружил характерные признаки белка и глюкозы в моче у ребенка с карликовым ростом и рахитом, то более популярно в медицине упоминание одного имени.
Болезнь чаще сопутствует другим наследственным патологиям обмена веществ (непереносимости фруктозы, галактоземии, тирозинемии). Распространенность составляет 1 случай на 350 тыс. родившихся живыми младенцев.
Причины
Современные генетические исследования установили роль изменений в 15-хромосоме (кодируется, как 15q15.3). Наследование мутантного гена может происходить по трем типам:
- аутосомно-рецессивному (необязательному);
- аутосомно-доминантному (обязательному);
- сцепленному с X-хромосомой.
Встречают и впервые произошедшие мутации у детей, не имеющих аналогичных изменений у родственников.
Клиницисты выделяют в зависимости от причины:
- первичный синдром Фанкони – это заболевание, сцепленное с Х-хромосомой, которое прогнозировать сложно из-за как рецессивного, так и доминантного пути наследования;
- вторичный – чаще сопровождает другие нарушения метаболизма.
Вторичное заболевание возникает:
- при наследственных изменениях почек;
- возможно развитие в результате трансплантации при недостаточной тканевой совместимости почки донора и пациента;
- отравлении ртутью, солями свинца, кадмия, урановыми соединениями;
- влиянии толуола, лизола и малеиновой кислоты на химических производствах;
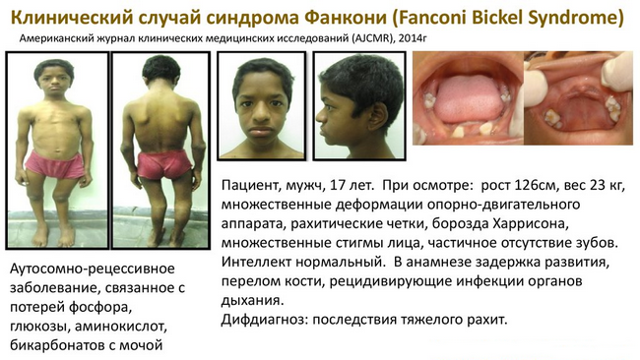
- терапии лекарственными препаратами на основе платины, антибиотиками Гентамицином и Тетрациклинам (особое значение имеет применение просроченных медикаментов).
Основная патология развивается в митохондриях клеток. Эти внутриклеточные структуры являются «фабрикой производства энергии» для всех видов деятельности. Для получения необходимых килокалорий в митохондриях происходит процесс фосфорилирования с участием кислорода.
Биохимическая реакция требует набора ферментов для поэтапных преобразований. Но при синдроме Фанкони в клетках почечного эпителия канальцев они отсутствуют. Следовательно, митохондрии не могут поставить необходимое количество энергии. Страдает реабсорбция в кровь необходимых веществ.
Вид митохондрии при электронной микроскопии, недостаточная выработка энергии лишает орган и ткань возможности функционировать
С мочой выводятся:
- глюкоза;
- альбумины;
- фосфаты;
- аминокислоты;
- бикарбонаты.
В крови регистрируется дефицит этих веществ и изменяется общий метаболизм:
- недостаток аминокислот и бикарбонатов приводит к сдвигу кислотно-щелочного равновесия в сторону закисления (ацидоза), при этом усиливается распад костной ткани;
- дополнительно снижается всасывание калия и кальция в почечных канальцах, элементы удаляются с мочой.
У ребенка в скелете появляются признаки рахита. Ближе к взрослому возрасту происходит процесс размягчения костной ткани – остеомаляция.
Симптомы болезни замечают у ребенка после 6-месячного возраста:
- дети мало двигаются;
- аппетит значительно снижен;
- мышцы развиваются плохо;
- постоянно просят пить;
- часто бывает рвота;
- повышается температура без связи с признаками инфекции;
- отстают в прибавке веса и физическом развитии;
- выделяют много мочи (полиурия);
- кожа сухая, обезвоженная.
Более ясная картина формируется на втором году жизни, а иногда в 5-6 лет. Здесь на первое место выступают:
- проявления деформации костной ткани, скелета;
- параличи, вызванные гипокалиемией.
Отставание в физическом и умственном развитии не вызывает сомнений. Ребенок растет пугливым и необщительным.
До года ребенок должен ежемесячно осматриваться педиатром
Костные изменения проявляются:
- в деформации ног;
- «утиной» походке;
- нарушении формы грудной клетки и позвоночника;
- измененного строения плечевых костей и предплечий;
- значительно сниженном мышечном тонусе.
Кости становятся ломкими. По этой причине у ребенка часто возникают переломы. Рост больного ниже сверстников.
По степени нарушения метаболизма и тяжести состояния пациента различают 2 клинических варианта синдрома Фанкони:
Синдром Альпорта у детей
- Первый – задержка физического и интеллектуального развития значительна, в течение заболевания преобладают тяжелые нарушения с выраженной гипокальциемией (до 1,6-1,8 ммоль/л), костными переломами и деформациями, снижается всасывание кальция не только в почечных канальцах, но и в кишечнике.
- Второй – отставание в физическом развитии менее выражено, умственно ребенок практически нормален, деформации костей незначительны, течение оценивается как легкое, в крови достаточная концентрация кальция, кишечник хорошо усваивает кальций.
Итогом болезни становится:
- патологические изменения нервной системы;
- ухудшение зрения;
- дефекты в развитии мочеполовых органов;
- хронические заболевания кишечника;
- состояние иммунодефицита;
- почечная недостаточность.
Проявление синдрома у взрослых
Во взрослом возрасте человек получает вторичный синдром Фанкони. Он проявляется:
- частым обильным мочеиспусканием (полиурией);
- жалобами на слабость;
- болями в костях;
- снижением тонуса мышц;
- склонностью к повторным переломам;
- изменения в почках приводят к стойкой гипертонии.
Тяжелее всего болезнь протекает у женщин в постклимактерическом периоде, когда добавляются гормональные влияния на баланс электролитов. Хрупкость костей приводит к тяжелым переломам:
- позвоночника;
- головки бедренной кости.
Это означает полную инвалидность, невозможность срастания костной ткани. Почечная недостаточность формируется постепенно. Эпителий клубочков атрофируется и замещается рубцовой тканью.
Компрессионный перелом позвонков опасен сдавлением спинного мозга
Диагностика
Выявление болезни основывается на рентгенологических и биохимических лабораторных методах. На рентгеновских снимках обнаруживают:
- разные деформации костей конечностей;
- истончение и атрофию коркового слоя в трубчатых костях;
- рыхлость в зонах роста;
- нарушение строения и формы позвоночника;
- плохо срастающиеся переломы;
- остеопороз всей костной ткани разной степени выраженности;
- отставание в темпе роста по возрасту ребёнка.
Среди биохимических нарушений выявляются в составе крови:
- снижение концентрации кальция и фосфора;
- рост фермента щелочной фосфатазы;
- гипокалиемию;
- избыток гормона паращитовидной железы;
- нарушение кислотно-щелочного баланса в сторону ацидоза.
В составе мочи:
- нормальное или повышенное выделение кальция;
- повышение содержания солей фосфатов;
- содержание глюкозы намного превышает почечный порог (20–30 г/л и выше);
- натрийурия;
- значительный уровень аминокислот.
Для точного диагноза необходимо отличить выявленные признаки от рахитоподобных заболеваний и осложнений при наследственных и других болезнях. Их отличают по дополнительному более полному исследованию крови, мочи, функции почек, костного мозга.
К подобным состояниям относятся:
- ювенильный нефронофтиз;
- галактоземия;
- цистиноз;
- тирозинемия;
- гликогенозы;
- непереносимость фруктозы;
- миеломная болезнь;
- гепатобилиарная дистрофия;
- амилоидоз;
- гиперфункция паращитовидных желез;
- последствия почечной трансплантации;
- синдром Шегрена;
- нефротический синдром,
- токсическое поражение почек при отравлениях солями тяжёлых металлов;
- передозировка лекарственных веществ, в том числе витамина D.
Дифференциальную диагностику проводит квалифицированный специалист
Как проводится лечение
Рекомендуется лечебная диета с увеличением фосфорсодержащих продуктов, аминокислот, витамина D. Полезны молочные продукты, свежие фрукты и соки. Дозировка согласуется с лабораторным контролем.
Лечение заключается:
- в возмещении потерь электролитов (калия, фосфатов, кальция);
- добавлении щелочи для поддержания кислотно-щелочного равновесия (в виде раствора бикарбоната натрия, цитратных смесей).
Врачами применяются:
- Аспаркам или Панангин ликвидируют потерю калия.
- Препараты кальция назначаются курсами.
- Показан общеукрепляющий массаж.
- Назначают препараты для стимуляции иммунитета.
- Курсы бальнеологической терапии ваннами из природных источников помогают укреплению костной ткани, мышц.
- При наличии почечной недостаточности показан гемодиализ с подбором состава диализата.
Для предотвращения необратимой патологии заболевание необходимо начинать лечить при первых проявлениях. Поэтому следует внимательно следить за развитием ребенка, необычной симптоматикой у взрослого человека.
Источник: https://sochi-mebel.ru/bolezni/sindrom-de-toni-debre-fankoni
Анемия Фанкони
Анемия Фанкони – это генетическое заболевание, которое передается по аутосомно-рецессивному типу и характеризуется нарушением кроветворения, формированием злокачественных новообразований, пороками развития, ломкостью хромосом. Проявляется частыми кровотечениями, кровоподтеками на коже, вялостью, бледностью, склонностью к инфекциям. Диагностика проводится лабораторными методами, назначаются цитогенетическое, молекулярно-генетическое и клиническое исследования крови, миелограмма. Основные способы лечения – пересадка костного мозга, медикаментозное поддержание кроветворения, переливание крови.
D61.0 Конституциональная апластическая анемия
Синонимичные названия анемии Фанкони – врожденная панмиелопатия Фанкони, наследственная панмиелопатия. Заболевание названо по фамилии швейцарского педиатра Гвидо Фанкони, который в 1927 году описал врожденную апластическую патологию на основе симптомов у трех братьев.
Анемия Фанкони является редкой генетической болезнью, наследуется согласно аутосомно-рецессивному принципу. Эпидемиологические показатели низкие – 1 больной ребенок на 350 тысяч новорожденных.
Распространенность одинакова среди представителей женского и мужского пола, выше в сообществах с разрешенными близкородственными браками, например, у некоторых южноафриканских народов.
Анемия Фанкони
Заболевание является наследственным, развивается при передаче дефектного гена от родителей к ребенку. Выявлено 15 генов, мутации которых проявляются анемией Фанкони. Из них 14 расположены в аутосомах и являются рецессивными, 1 тип гена находится в X-хромосоме (сцепленной с полом). Все эти гены отвечают за производство определенного фермента, участвующего в репарации ДНК.
Аутосомно-рецессивное наследование подразумевает, что и отец, и мать должны быть носителями патологической генетической информации. При этом сами они, как правило, здоровы. Вероятность рождения больного ребенка в такой паре составляет 25%.
Генетическая панмиелопатия диагностируется у детей и взрослых, получивших от каждого из родителей один и тот же измененный ген. В крайне редких случаях анемия провоцируется передачей дефектной Х-сцепленной хромосомы. Женщины могут быть носительницами мутации, заболевание проявляется только у мальчиков.
Риск развития патологии у сына при наличии у матери мутированного гена – 50%.
В норме в клетках организма существуют специальные ферментные системы, которые исправляют разрывы молекул ДНК, поврежденных в процессе биосинтеза или воздействия химических, физических реагентов.
При анемии Фанкони обнаруживается генетический дефект в кластере белков, ответственных за репарацию ДНК, что приводит к повышенной ломкости хромосом. В итоге у пациентов развиваются нарушения функций костного мозга – неоплазии и апластическая анемия.
Онкологические заболевания чаще всего представлены острым миелоидным лейкозом – злокачественной опухолью миелоидного ростка крови, провоцирующей накопление измененных белых клеток, подавляющих рост эритроцитов, тромбоцитов и нормальных лейкоцитов.
При апластической анемии в результате дисплазии костного мозга резко угнетается рост и созревание всех трех видов клеток крови.
Более чем у половины пациентов наблюдаются врожденные аномалии развития внутренних органов и скелета.
Костные деформации проявляются специфическим внешним видом: больные низкорослые, с уменьшенным размером головы, отсутствием или заметным укорочением большого пальца на руках, недоразвитием лучевой кости, врожденным вывихом бедра и/или наличием шейного ребра, косолапостью, недоразвитым подбородком («птичьим лицом»). Характерна гиперпигментация кожи в виде светлых и коричневатых пятен.
Неврологические расстройства представлены косоглазием, недоразвитием одного или двух глаз, опущением верхнего века, глазным дрожанием, глухотой, умственной отсталостью. Больные зачастую имеют незрелые половые органы, у них отсутствует одно или оба яичка.
К распространенным аномалиям строения органов относятся пороки мочевыделительной системы: удвоение мочеточников или лоханки, подковообразные почки, почечные кисты, смещенное наружное отверстие уретры (гипоспадия).
Врожденные пороки сердца включают атрезию трехстворчатого клапана, дефект межпредсердной перегородки, митральный стеноз, дефект межжелудочковой перегородки. Пациенты страдают от почечной и сердечной недостаточности.
Ключевые симптомы связаны с постепенным нарастанием нарушений в работе костного мозга. Чаще они дебютируют в детском возрасте (в 5-10 лет).
Из-за снижения количества тромбоцитов развивается повышенная кровоточивость: при ранениях кровь долго не сворачивается, легко возникают носовые кровотечения, выделения при менструациях обильны, на теле обнаруживается много «беспричинных» кровоподтеков.
Уменьшение числа эритроцитов проявляется анемией с характерной слабостью, быстрой утомляемостью, головокружениями, обмороками, бледностью кожи, учащенным сердцебиением и одышкой. Недостаток лейкоцитов способствует ухудшению сопротивляемости инфекциям. Впоследствии формируется лейкоз, миелодиспластический синдром, онкологические болезни.
Наиболее распространенным осложнением считаются частые инфекционные заболевания. У пациентов развивается ОРВИ, ангина, ринит, бронхит, грипп, тиф, герпес. Рецидивирующий характер болезней и их тяжелое течение приводят к деструкции органов, сопровождаются риском сепсиса.
Другим осложнением наследственной анемии являются злокачественные новообразования – лейкемия, эпителиальные опухоли органов шеи и головы, половых органов. Рак у таких больных тяжело поддается лечению из-за повышенной ломкости и сниженной репарации ДНК. Это явление ограничивает применение лучевой терапии, цитотоксических препаратов.
Нарушение свертываемости становится причиной больших кровопотерь.
Обследование больных проводят онкологи, гематологи, педиатры, врачи-генетики. Диагностика начинается с анализа анамнестических данных и жалоб. Врач выясняет, имеется ли данное наследственное заболевание у близких родственников, уточняет время появления первых признаков болезни, ранние обращения к врачам.
При осмотре оценивает общее состояние пациента, выявляет наличие аномалий развития, гиперпигментированных пятен, кровотечений, кровоподтеков. В большинстве случаев не составляет труда обнаружить типичные деформации костей, недоразвитие глаз.
Для подтверждения диагноза и различения анемии Фанкони с приобретенной анапластической анемией проводится ряд лабораторных исследований:
- Клинический анализ крови. Характерны изменения клеточного состава крови. На ранних этапах нарушения кроветворения диагностируется тромбоцитопения и лейкопения, на более поздних – панцитопения (резкое снижение объема эритроцитов, лейкоцитов и тромбоцитов). Возможен умеренный гемолиз без гипербилирубинемии, но с ретикулоцитозом. Значение СОЭ увеличено до 60-80 мм/ч.
- Цитогенетическое исследование клеток. Выполняется проба с диэпоксибутаном, митомицином C, указывающая на частоту и спектр хромосомных аберраций. В пользу генетической анемии рассматриваются показатели ДЭБ-теста более 45%, пограничный уровень – 11-45% (процент клеток с хромосомными разрывами).
- Молекулярно-генетический анализ клеток. Исследуются гены, мутации в которых могут привести к развитию заболевания. В 60-70% случаев мутации обнаруживаются в паре генов FANCA, в 14% – в аллели FANCC, в 10% – в генах FANCG. Частота мутаций в других парах – 0,2-3%.
- Миелограмма. По данным исследования определяется увеличение количества плазматических клеток и макрофагов, фагоцитирующих жиры. Содержание недифференцированных клеток – в пределах нормы. Снижена концентрация клеток миелоцитарного ростка, увеличен показатель лимфоцитов.
Основная терапия направлена на восстановление процесса кроветворения. Методы лечения подбираются индивидуально, зависят от тяжести заболевания, возраста пациента, наличия и выраженности врожденных аномалий. Дополнительно проводится лечение инфекций и онкопатологий, осуществляются реабилитационные мероприятия. Для устранения анемии используются следующие методы:
- ТКМ. Трансплантация костного мозга является наиболее эффективной в долгосрочной перспективе, но имеет противопоказания, нередко сопровождается развитием осложнений. Оптимальный возраст для проведения операции – до десяти лет. Донорами могут выступать здоровые сестра и братья, подходящие по критериям совместимости. Предварительная интенсивная терапия (кондиционирование) связана с риском токсического воздействия на органы. После трансплантации сохраняется высокая вероятность острого или хронического иммунного конфликта между клетками донора и реципиента.
- Медикаментозная стимуляция кроветворения. При невозможности проведения трансплантации пациентам показано консервативное лечение, временно улучшающее их состояние. Выработка кровяных клеток стимулируется андрогенами (мужскими половыми гормонами) и гематопоэтическими факторами роста – эритропоэтином, фактором стволовых клеток, интерлейкинами-1-12. Параллельно применяются иммунодепрессанты. Медикаментозная терапия способна на протяжении многих лет поддерживать высокое качество жизни больных, но ее эффективность постепенно снижается.
- Переливание компонентов крови. При выраженных побочных эффектах или противопоказаниях к этиотропной терапии (трансплантации, стимуляции кроветворения) назначаются процедуры гемотрансфузии. Переливаются отмытые эритроциты – донорские красные кровяные тельца, освобожденные от поверхностных белков. При кровотечениях и снижении уровня тромбоцитов пациентам вводится тромбоцитарная масса.
Продолжительность жизни больных определяется степенью нарушения функции костного мозга. Иногда пациенты доживают до 40 лет без лечения, но нередко умирают в детстве от тяжелой анемии или онкологических заболеваний.
Прогноз наиболее благоприятен при своевременном проведении аллогенной трансплантации костного мозга, после которой есть шанс полного восстановления нормального кроветворения и увеличения срока жизни. Поскольку заболевание генетическое, предотвратить его развитие невозможно.
Профилактика сводится к медико-генетической консультации супружеских пар из групп риска, планирующих беременность, а также к проведению пренатальной диагностики патологии, в ходе которой из пуповинной вены плода производится забор крови и выполняется ДЭБ-тест.
При его положительном результате рассматривается вопрос о прерывании беременности.
Источник: https://www.KrasotaiMedicina.ru/diseases/genetic/Fanconi-anemia
Синдром фанкони у детей говорит о мутации генов. Болезнь де Тони-Дебре-Фанкони: причины, симптомы, принципы питания и лечения у детей Синдром Фанкони у животных
Болезнью де Тони-Дебре-Фанкони называют врожденное, генетически обусловленное заболевание, приводящее к выраженному нарушению функции почечных канальцев. Вследствие этого происходят потери с мочой глюкозы, фосфатов, аминокислот, снижается концентрация гидрокарбонатов и нарушается кислотно-щелочной баланс.
Эта наследственная патология встречается очень редко: у 1 больного младенца из 350 тысяч родившихся деток вне зависимости от пола новорожденного.
Причины
Причина данной патологии кроется в мутации определенных генов, однако что именно приводит к этой мутации ученым выяснить пока не удалось.
И характер, и причины синдрома Фанкони недостаточно изучены. Пока специалистами не установлено, биохимический или структурный дефект является основой болезни.
Предполагают, что причиной генетического порока может быть мутация генов, обуславливающая функциональные нарушения ферментов, которые участвуют в регуляции всасывания в почечных канальцах аминокислот, глюкозы и фосфора.
Трактовка этой патологии среди ученых неоднозначна. Одни из них рассматривают ее как самостоятельное наиболее тяжелое рахитоподобное заболевание. Потеря важных веществ приводит к развитию дистрофических изменений в костной ткани.
Рахитоподобные изменения возникают при сочетании дефицита фосфатов с ацидозом (кислой реакцией крови), хотя не все ученые разделяют это мнение. Предполагают, что сниженная чувствительность при этом к витамину D связана с невозможностью его превращения в активную форму при ацидозе.
Другие считают патологию приобретенным синдромом, связанным с целым рядом состояний:
- болезнью Коновалова-Вильсона;
- многими нарушениями ферментативных систем;
- непереносимостью фруктозы;
- токсическим действием тяжелых металлов или некоторых лекарственных средств;
- дефицитом витамина D;
- следствием некоторых приобретенных заболеваний: амилоидоза, злокачественных заболеваний, множественной миеломы и др.
Так как единого мнения о причинах патологии нет, то для ее обозначения пользуются различной терминологией: «наследственный синдром Фанкони», «D-резистентный рахит», «глюкофосфаминный диабет» и др.
Различают полный синдром Фанкони, при котором организм теряет глюкозу, аминокислоты и фосфаты, и неполный – с двумя из этих биохимических дефектов.
Наследственный вид генетики связывают с возникновением дефекта в Х-хромосоме. Наследование мутации происходит и по рецессивному, и по доминантному типу, что значительно затрудняет генетическое прогнозирование для потомства.
Врожденный синдром Фанкони проявляется на первом году жизни. Как правило, наследственная форма болезни Фанкони сопровождается и другой врожденной патологией.
Среди медикаментозных средств, способных вызвать приобретенный синдром Фанкони, можно назвать нефротоксичные:
- препараты для химиотерапии при злокачественных опухолях (Стрептозоцин, Ифосфамид);
- Тетрациклин с истекшим сроком годности;
- Гентамицин;
- препараты платины;
- противовирусные препараты (Цидофовир, Диданозин).
Приобретенный синдром Фанкони может возникать при пересадке почки с недостаточной совместимостью, глубоких ожогах и ряде других заболеваний.
Симптомы
Первые симптомы наследственного заболевания появляются в течение первых месяцев после рождения малыша. В более редких случаях они отмечаются после 1,5 лет.
Признаками врожденного синдрома Фанкони у грудничков являются:
- учащенное мочеиспускание;
- выраженная жажда;
- часто возникающая беспричинная рвота;
- упорные ;
- вздутие живота;
- быстрая утомляемость;
- беспричинные подъемы температуры в пределах 37,5- 38 0 С;
- мышечная слабость.
При этом нет проявлений, характерных для энтеровирусной инфекции или ОРВИ.
Если симптомы у грудничка были неопределенными или слабо выраженными, то в последующие 1-1,5 года проявления синдрома Фанкони становятся более явными:
- Появляется отставание ребенка в росте и весе от нормативных по возрасту: дефицит веса составляет около 30 %, а роста – от 2 до 21 %. Ранний нанизм (низкий рост) связан с постоянными потерями организмом аминокислот, фосфатов, глюкозы, кальция.
- Проявления рахита видны уже с возраста 10-12 месяцев. Отличительными особенностями рахита при синдроме Фанкони являются выраженная деформация и искривления костей конечностей, грудной клетки, позвоночника при незначительных изменениях головки малыша.
Кости нижних конечностей могут деформироваться по вальгусному типу (искривление голеней имеет вид буквы «Х») или по варусному типу (голени искривлены «колесом»).
- Ребенок отстает не только в физическом, а и в умственном развитии. В поведении характерны пугливость, необщительность.
- Потери натрия приводят к нарушению обратного всасывания воды. Жажда и повышение выделения мочи за сутки, учащение мочеиспусканий могут то ослабевать, то усиливаться, но полностью не исчезают.
- Снижение мышечного тонуса приводят к затруднениям движений, вследствие чего малыши даже к 5-6 годам не могут ходить. Если все же дитя начинает самостоятельно передвигаться, то походка у него неуверенная, «утиная».
- Умеренно выраженные или сильные болевые ощущения в костях конечностей, позвоночнике, костях таза могут затруднять передвижение ребенка.
- Дефицит минералов фосфора и кальция повышает риск возникновения переломов трубчатых костей и размягчения костей.
- Из-за значительных потерь глюкозы и бикарбонатов происходят большие потери калия. Дефицит калия в организме повышает вероятность возникновения параличей и нарушений со стороны сердца.
Выраженный дефицит калия в организме имеет такие клинические проявления: слабость, тошнота, учащение сердцебиений, изменения на ЭКГ, снижение тонуса мышц и рефлексов, расширенные границы сердца.
Потеря гидрокарбонатов ведет к ацидозу, который проявляется бледностью кожи и слизистых, раздражительностью, слабостью.
- Развитие офтальмологической патологии (катаракты, пигментного ретинита).
- Возможно поражение нервной и сердечно-сосудистой систем, органов пищеварения вследствие выраженных нарушений обменных процессов.
- В редких случаях возникают эндокринные нарушения.
- Снижение иммунитета приводит к частым ОРВИ, пневмонии, отитам.
При прогрессирующем развитии болезни уже к 10-12 годам создается высокий риск развития ХПН (хронической почечной недостаточности), создающей угрозу для жизни.
Диагностика
Среди методов диагностики синдрома Фанкони большое значение имеют общий и биохимический анализы крови.
 Среди методов диагностики синдрома Фанкони большое значение имеют общий и биохимический анализы крови.
Среди методов диагностики синдрома Фанкони большое значение имеют общий и биохимический анализы крови. Для диагностики заболевания используют:
- глубокое биохимическое исследование крови и мочи;
- общий анализ мочи и крови;
- биопсию ткани почки;
- рентгенологическое исследование костей конечностей и позвоночника для обнаружения деформаций и уменьшения зоны роста костей;
- УЗИ почек.
Может понадобиться консультация офтальмолога, ортопеда, уролога, нефролога, генетика.
Лечение
Правильно назначенное лечение дает возможность уменьшить негативное влияние значительных потерь минералов, аминокислот, глюкозы на костную систему, головной мозг и другие органы. Госпитализация показана при наличии резких обменных нарушений и деформаций скелета.
Применяемая терапия направлена на:
- максимально возможную коррекцию кислотно-щелочного баланса, дефицита бикарбонатов и калия;
- лечение D-резистентного рахита (без ограничения потребляемой жидкости).
Медикаментозное лечение включает: назначение специальных препаратов витамина D3 с индивидуальным подбором дозы ребенку под контролем уровня фосфора в крови. Препарат назначается несколькими курсами для предотвращения прогрессирования костных деформаций.
Могут использоваться активные метаболиты D3 – Рокальцитрол и Оксидевит, препараты фосфора, кальция, фитин. Неорганические фосфаты могут применяться в виде раствора (смесь Олбрайта) или таблеток в индивидуально подобранной дозировке (обязательно с одновременным лечением витамином D, чтобы не допустить гиперпаратиреоза, то есть повышения функции паращитовидной железы).
В случае выраженного дефицита калия назначаются Аспаркам, Панангин.
Регулярно контролируется реакция крови (в норме она слабощелочная, рН 7,35-7,45). Если рН ниже 7,35, необходимы меры по ощелачиванию кислой крови. С этой целью может назначаться вливание в вену 4% р-ра гидрокарбоната натрия.
Может применяться ощелачивающая микстура (состав: 100 мл воды, 2 г лимонной кислоты, 3 г цитрата натрия, 3,3 г цитрата калия) для приема внутрь по 50 мл за день, или же для ощелачивания применяют питьевую соду.
При значительных потерях цистина (аминокислоты) назначаются Дитиотрентал, Цистеамин. Пеницилламин способствует уменьшению в крови содержания пировиноградной кислоты и обеспечению резерва щелочи, уменьшает он также потери аминокислот.
Анаболики (Метилтестостерон) способны улучшить функциональную способность канальцев почек. Положительный результат получают и при назначении Унитиола.
После нормализации фосфорно-кальциевого обмена и устранения ацидоза могут применяться ванны (с морской солью, хвоей), массаж. Они окажут благотворное действие на организм ребенка.
Хирургическое лечение проводится в случае выраженной деформации костей. Оперативная коррекция может проводиться при достижении стойкой ремиссии, длящейся 1-1,5 года, при ее подтверждении лабораторными результатами.
В случае развития ХПН спасти жизнь ребенку может пересадка почки.
Диетотерапия
В рационе страдающего данной патологией ребенка обязательно должны присутствовать молоко и прочие молочные продукты.
 В рационе страдающего данной патологией ребенка обязательно должны присутствовать молоко и прочие молочные продукты.
В рационе страдающего данной патологией ребенка обязательно должны присутствовать молоко и прочие молочные продукты. При синдроме Фанкони соблюдение диеты – это один из основных компонентов лечения. Цель лечебного питания – обеспечение нормализации содержания в сыворотке крови фосфора, калия, ограничение потерь серосодержащих аминокислот, активация процессов окостенения, устранение ацидоза. Жидкость и белки не ограничиваются, а вот потребление соли желательно уменьшить.моркови , кураги, абрикосов.
С целью уменьшения задержки умственного развития ребенок при высоких показателях сахара в моче должен получать в достаточном количестве сахар и сладкий десерт. Однако чрезмерное употребление сладостей тоже не следует допускать, так как при избыточном уровне глюкозы в крови увеличится он и в моче.
Значительное содержание серосодержащих аминокислот отмечается в таких продуктах:
- крабы;
- рыба;
- миндаль и арахис;
- овес и зародыши пшеницы;
- чечевица и фасоль;
- кукуруза;
- семена кунжута;
- крупы (гречка, пшено, перловая, манная, коричневый рис).
Следует помнить, что чрезмерное количество серосодержащих аминокислот вредно для организма. А так как в некоторых продуктах содержатся и эти аминокислоты, и нужные организму фосфаты, то диету необходимо согласовывать с врачом. Лучше всего, если рацион подберет диетолог.
Прогноз
Прогноз зависит от:
- формы синдрома Фанкони;
- выраженности проявлений;
- сроков начала лечения.
- При развитии вторичного синдрома все его проявления исчезнут после устранения основной патологии или уменьшатся при активном лечении болезни, послужившей причиной появления синдрома Фанкони.
- При врожденной форме синдрома известны случаи продолжительной ремиссии и даже излечения.
- Угроза жизни создается при развитии ХПН.
Резюме для родителей
Синдром Де Тони-Дебре-Фанкони, к счастью, редко встречается у детей. В случае его развития следует начать лечение в ранние сроки, чтобы предупредить возможные тяжелые последствия патологии – отставание в развитии, как физическом, так и умственном, грубые деформации костей.
Генетическое консультирование перед планируемой беременностью (если случаи болезни были у родственников) поможет предупредить рождение больного ребенка.
Познавательное видео о синдроме Фанкони (англ. яз.):
Нажав на кнопку «Скачать архив», вы скачаете нужный вам файл совершенно бесплатно.
Перед скачиванием данного файла вспомните о тех хороших рефератах, контрольных, курсовых, дипломных работах, статьях и других документах, которые лежат невостребованными в вашем компьютере.
Это ваш труд, он должен участвовать в развитии общества и приносить пользу людям. Найдите эти работы и отправьте в базу знаний.
Мы и все студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будем вам очень благодарны.
Чтобы скачать архив с документом, в поле, расположенное ниже, впишите пятизначное число и нажмите кнопку «Скачать архив»
- Понятие и клиническая картина, а также причины и факторы развития болезни Дебре-де Тони-Фанкоии как наследственного заболевания, передающегося по аутосомно-рецессивному типу. Этиология и патогенез болезни, принципы ее лечения, прогноз на выздоровление.презентация , добавлен 02.12.2014 Факторы формирования тубулопатий. Болезни, которые относятся к рахитоподобным. Причины развития Витамин-D-резистентного рахита, его диагностика и лечение. Болезнь де Тони-Дебре-Фанкони (глюкоаминофосфат-диабет), ее клиническая картина и методы лечение.
Источник: https://duyc.ru/sindrom-fankoni-u-detei-govorit-o-mutacii-genov-bolezn-de-toni-debre-fankoni.html
Синдром Фанкони
Синдром Фанкони (глюкозо-фосфат-аминовый диабет, болезнь де Тони-Дебре-Фанкони, первичный изолированный синдром Фанкони) — генетическое заболевание, развившееся в результате аутосомно-рецессивной мутации, характеризующееся нарушением обратного всасывания воды и биоактивных веществ из первичной мочи (канальцевая реабсорбция), обусловленное поражением почечных канальцев. Относится к рахитоподобной группе заболеваний, при которых происходят системные метаболические изменения.
Онлайн консультация по заболеванию «Синдром Фанкони».
Задайте бесплатно вопрос специалистам: Эндокринолог.
Патологические изменения представляют одну из форм гиперпаратиреоза — эндокринного расстройства, развивающегося при избыточной выработке паратгормона (производится паращитовидными железами) в результате гиперплазии желез или злокачественного поражения.
Варианты наследования синдрома де Тони-Дебре-Фанкони:
- Аутосомно-доминантный — дефектный ген наследуется от одного из родителей (семейная форма).
- Аутосомно-рецессивный — дефектный ген присутствует у обоих родителей. В случае с синдромом речь идет о локальной форме аутосомно-рецессивного наследования (хромосома 15q15.3.)
Синдром Фанкони у детей может быть компонентом других генетических заболеваний:
- Цистиноз — чрезмерное накопление цистина (аминокислота) в цитоплазме (внутренняя жидкая клеточная среда) клетки.
- Галактоземия — нарушение преобразования галактозы (моносахарид) в глюкозу, обусловленное мутацией гена, отвечающего за выработку галактозо-1-фосфатуридилтрансферазы (фермент).
- Тирозинемия I типа — недостаток фумарилацетоацетата гидролазы, приводящей к нарушению метаболизма тирозина.
- Болезнь Вильсона — тяжелая гепатоцеребральная дистрофия, обусловленная нарушением метаболизма меди.
- Мальабсорбция фруктозы — потеря фермента в результате нарушения всасывания фруктозы, ее непереносимость, обусловленная дефицитом белка-транспортера фруктозы.
Установлено, что в основе патологии лежит комбинированная тубулопатия — группа болезней, при которых нарушен транспорт биологически активных веществ в канальцевой системе. Главное звено механизма развития синдрома Фанкони — дефект митохондрий (энергетическое депо клетки) в цикле трикарбоновых кислот (цикл Кребса), являющейся ключевой стадией дыхания клеток.
Этапы механизма развития болезни, где каждый последующий этап будет следствием предыдущего, можно представить следующим образом:
- Митохондриальный дефект, ферментная тубулопатия.
- Нарушение реабсорбции аминокислот и ферментов в канальцах почек.
- Накопление кислот (метаболический ацидоз).
- Костная резорбция (разрушение).
- Нарушение обратного всасывания кальция и калия в канальцах.
- Гипокалиемия, гиперкальциемия.
Клетки теряют энергообеспечение, в результате чего развиваются тяжелые нарушения обмена веществ. Среди факторов риска выделяют:
- отравление тяжелыми металлами;
- токсикоинфекции;
- прием просроченных антибиотиков тетрациклинового ряда;
- дефицит витамина D;
- амилоидоз (нарушение белкового обмена).
Выделяют первичную (идиопатическую) и вторичную формы болезни Фанкони. Первичная форма развивается в результате наследования дефектного гена. Вторичная форма возникает при других врожденных, генетически обусловленных заболеваниях.
Вторичный синдром может возникать на фоне приобретенных патологий:
- парапротеинемия — наличие в крови аномальных белковых тел;
- нефротический синдром — тяжелые нарушения, развивающиеся при поражении клубочков почек;
- тубулоинтерстициальные нефропатии — группа болезней почек, характеризующихся первичным поражением канальцев;
- злокачественное новообразование (паранеопластический синдром);
- интоксикация при отравлениях;
- ожоги тяжелой степени.
Симптомы синдрома Фанкони
У синдрома Фанкони, симптомы которого появляются у детей ко второму году жизни, имеется два варианта развития, в зависимости от клинико-лабораторных показателей:
- Первый вариант — характерно тяжелое течение, выраженное отставание в физическом развитии, переломы и деформации костей в результате гипокальциемии, нарушение всасывания в кишечнике.
- Второй вариант — относительно легкое течение, умеренные признаки задержки в физическом развитии, деформации костей при нормальном уровне кальция, всасывание кальция в кишечнике в пределах нормы.
Первые симптомы болезни у детей до двух лет:
- резкое снижение аппетита;
- дефицит массы тела;
- вялость;
- гипотрофия (нарушение пищеварения, обусловленное белково-энергетической недостаточностью);
- жажда;
- низкое артериальное давление;
- полиурия (большое количество мочи);
- субфебрильная температура;
- запоры;
- рвота.
Такие дети примерно к пяти годам не могут ходить.
При разгаре заболевания к пяти-шести годам первым признаком будет выступать остеомаляция (костное размягчение), костные деформации, параличи, связанные с недостатком кальция.
После появления первых признаков наблюдается отставание в умственном и физическом развитии.
Генерализованная (распространенная на весь организм) декальцификация проявляется деформациями нижних конечностей (вальгусная деформация — искривление вовнутрь, варусная — искривление вовне), потеря мышцами тонуса, искривление грудной клетки, костей предплечий и плеч. Нехватка фосфора у детей приводит к появлению рахита.
При прогрессировании синдрома у детей выявляются зрительные расстройства, болезни нервной системы, мочевой и пищеварительной системы, болезни ЛОР-органов. Редко возникают иммунодефицитные состояния.
При синдроме Фанкони у взрослых происходит остеомаляция, обусловленная дефицитом минералов и микроэлементов. Пациенты жалуются на боли в костях, слабость в мышцах, вялость, возможно повышение артериального давления, развитие почечной недостаточности при отсутствии терапии.
У детей раннего возраста даже на первых неделях жизни могут возникнуть признаки синдрома Висслера-Фанкони, обусловленные выраженной аллергической реакцией. Характеризуется патология лихорадкой, эритематозной сыпью, поражением суставов (чаще рук).
Для выявления болезни Фанкони используют лабораторные и визуальные методы диагностики. Биохимическое исследование крови позволяет выявить недостаток кальция и фосфора, бета-2-микроглобулин (низкомелекулярный белок).
В моче выявляется аминоацидурия (продукты обмена аминокислот), почечный ацидоз (электролитные нарушения), гликозурия (сахар в урине), большое количество фосфатов, дефицит микроэлементов (натрия, кальция, калия, фосфора и других).
В оценке функции почек большую роль играет ультразвуковое обследование и МРТ.
Рентгенологическое исследование костей позволяет изучить костную структуру, обнаружить нарушения структуры, степень остеопороза, деформацию, оценить возрастное отставание развития костной ткани. При болезни Фанкони с помощью рентгенографии обнаруживают:
- грубоволокнистую структуру кости;
- эпифизеолиз — разрушение эпифизарной (хрящевой) пластинки роста;
- ячеистую структуру и шпороподобные наросты в большеберцовых костях;
- остеопороз и переломы — на поздних стадиях.
- При позитронно-эмиссионном исследовании (ПЭТ) выявляют накопление радиоизотопного вещества в зонах роста костей больного.
- В биопсийном материале находят нарушение структуры кости, лакуны (патологические углубления), плохую минерализацию.
- При глюкозо-фосфат-аминовом диабете проводится дифференциальная диагностика с такими патологиями:
- цистиноз;
- гликогеноз;
- разные синдромы (Лоу, Шегрена, нефротический);
- множественная миелома (злокачественная болезнь крови);
- непереносимость фруктозы, обусловленная генетическим фактором, другими наследственными заболеваниями;
- сахарный диабет;
- состояния, возникающие при трансплантации почки.
Лечением синдрома Фанкони занимается гематолог и генетик. При артериальной гипертензии, аномалиях строения почечных структур, выраженной протеинурии необходима консультация уролога и нефролога, при эндокринных расстройствах — эндокринолога, при нарушениях зрения — офтальмолога.
Терапия направлена на:
- устранение электролитных нарушений, в частности тубулярного ацидоза;
- коррекцию кислотно-щелочного дисбаланса;
- устранение клинических проявлений (симптоматическая терапия).
Курсом назначаются препараты калия и кальция, витамин D c постепенным увеличением дозировки и исследованием крови в динамике на содержание фосфора и кальция. Рекомендовано обильное питье и диета.
В питании следует ограничить потребление соленых продуктов и соли, ввести в рацион молоко, курагу, чернослив, фруктовые соки.
При нормализации показателей крови можно делать массаж, принимать хвойные ванны.
Хирургическое вмешательство показано только при выраженных деформациях костей. Выполняется операция при стойкой ремиссии продолжительностью от полутора лет, которая подтверждена диагностическими показателями и клиническими проявлениями.
Самое тяжелое осложнение глюкозо-фосфат-аминового диабета — почечная недостаточность. При тяжелой степени заболевания, угрожающей для жизни пациента, показан гемодиализ («искусственная почка»).
Процедура гемодиализа
При некоторых видах почечной недостаточности гемодиализ проводится временно, до улучшения или восстановления почечной функции. В остальных случаях, при необратимых процессах в почках, процедура проводится пожизненно.
Диализ заключается в пропускании крови по специальной системе, где из биожидкости отделяются токсические вещества, которые удаляются с помощью диализирующего раствора. Организм освобождается от отравляющих продуктов распада до следующего накопления.
Прогноз зависит от основного заболевания, на фоне которого развился синдром. Если основная болезнь — новообразование, при успешном его удалении прогноз может быть относительно благоприятным.
Болезнь Фанкони вызывает развитие сложных последствий, которые приводят к инвалидизации пациента. Летальный исход наступает при почечной недостаточности, когда гемодиализ уже не оказывает должного эффекта, а трансплантация почки не проведена. Либо летальный исход может наступить при неудачной трансплантации органа.
Основная профилактика заболевания — своевременный генетический скрининг при отягощенном анамнезе. Если в семье есть или были родственники с генетическими расстройствами, обязательно нужно проходить диагностику с целью выявления возможных рисков. Для сестер и братьев риск заболеть синдромом при отягощенном анамнезе составляет 25 %.
Источник: https://SimptoMer.ru/bolezni/endokrinnaya-sistema/3433-sindrom-fankoni
Болезнь де Тони — Дебре — Фанкони
Болезнь де Тони — Дебре — Фанкони
(глюкоаминофосфат-диабет) представляет
собой наиболее тяжелое заболевание
среди Р. б. Наследуется по аутосомно-рецессивному
типу, однако экспрессивность мутантного
гена в гомозиготном состоянии значительно
варьирует; встречаются спорадические
случаи, обусловленные свежей мутацией.
Полагают, что в основе болезнилежат
генетически обусловленные дефекты
ферментативного фосфорилирования в
почечных канальцах (комбинированная
тубулопатия).
Это приводит к нарушению
процессов энергообеспечения транспорта
фосфатов, глюкозы и аминокислот в
почечных канальцах и повышенной их
экскреции с мочой, а также расстройству
механизмов поддержания равновесия
кислот и оснований.
Развивающийся
метаболический ацидоз и недостаток
фосфорных соединений способствуют
нарушению формирования костной ткани
по типу остеомаляции и рахитоподобных
изменений скелета. В ряде случаев
выявляют морфологические изменения в
почечных канальцах, нарушение функции
паращитовидных желез, расстройства
синтеза 1,25-диоксихолекальциферола.
В большинстве случаев первые признаки
заболеванияпоявляются во второй
половине первого года жизни, развернутый
симптомокомплекс формируется ко второму
году жизни; реже наблюдается поздняя
манифестация болезни — в 6-7-летнем
возрасте. Начальные клинические
проявления — повышенная жажда, полиурия,
иногда длительный субфебрилитет, рвота.
На втором году жизни выявляют отставание
физического развития и костные
деформациинижних конечностей (вальгусные
или варусные), грудной клетки, предплечий
и плечевых костей (рис. 3).
Рентгенологически
при этом определяют остеопороз системный
различной степени выраженности,
истончение коркового слоя трубчатых
костей, разрыхление зон роста, отставание
темпов роста костной ткани от биологического
возраста ребенка.
Характерными особенностямибиохимических нарушений при болезни
де Тони — Дебре — Фанкони являются снижение
уровня кальция и фосфора в крови,
повышение активности щелочной фосфатазы,
метаболический ацидоз (рН — 7,35-7,25; ВЕ =
-10-12 ммоль/л).
Экскреция кальция с мочой
обычно остается нормальной при повышенном
клиренсе фосфатов мочи. Отмечают
глюкозурию (20-30 г/л и выше), генерализованную
гипераминоацидурию и нарушение функций
аммониоацидогенеза — снижение титрационной
кислотности, повышение рН мочи > 6,0.
При полиурии до 2 л и более в сутки
удельная плотность мочи, как правило,
высокая (1025-1035), что обычно связано с
глюкозурией.
В зависимости от тяжести клинических
проявлений и метаболических расстройств
выделяют два клинико-биохимических
вариантаболезни де Тони — Дебре —
Фанкони.
Первый характеризуется
значительной задержкой физического
развития, тяжелым течением заболевания
с выраженными костными деформациями и
нередко переломами костей, резкой
гипокальциемией (1,6-1,8 ммоль/л), снижением
абсорбции кальция в кишечнике.
При
втором варианте отмечают умеренную
задержку физического развития, легкое
течение с незначительными костными
деформациями, нормокальциемию и
нормальное усвоение кальция в кишечнике.
Критериями диагностикиявляются
выраженный дефицит массы тела и роста
ребенка, задержка становления
статико-моторных функций, рахитоподобные
деформации скелета с характерной
рентгенологической картиной нарушений
структуры костной ткани, отмеченные
особенности электролитных нарушений.
Дифференциальный диагнозпроводят
с рахитом, остеопатиями вследствие
хронической почечной недостаточности;
кроме того, большое значение имеет
дифференцирование первичной болезни
де Тони — Дебре — Фанкони с вторичным
синдромом, обнаруживаемым при других
наследственных и приобретенных
заболеваниях (синдроме Лоу, ювенильном
нефронофтизе, цистинозе, тирозинемии,
галактоземии, гликогенозах, наследственной
непереносимости фруктозы, гепатоцеребральной
дистрофии, миеломной болезни, амилоидозе,
синдроме Шегрена, нефротическом синдроме,
при почечной трансплантации,
гиперпаратиреозе, поражении почек
солями тяжелых металлов, отравлении
лекарственными веществами, в т. ч.
витамином D, лизолом и т.д.). Синдром Лоу
(окулоцереброренальный синдром) в
отличие от болезни де Тони — Дебре —
Фанкони характеризуется отставанием
в умственном развитии, двусторонней
катарактой, глаукомой, гипорефлексией,
а также рецессивным, связанным с полом
типом наследования. Ювенильный нефронофтиз
Фанкони, морфологической основой
которого являются кисты в мозговом
веществе почек на уровне собирательных
трубочек и гиалиноз почечных клубочков,
отличается ранним нарушением
концентрационной функции почек
(гипостенурией), нормохромной анемией,
гиперазотемией; рахитоподобные изменения
скелета присоединяются позже. Решающую
роль в диагностике нефронофтиза Фанкони
играет исследование биоптата почечной
ткани.
Основные принципы лечениязаключаются
в коррекции электролитных нарушений,
сдвигов в кислотно-щелочном равновесии,
устранении дефицита калия и бикарбонатов.
Особенности применения витамина D и его
метаболитов для ликвидации нарушений
фосфорно-кальциевого гомеостаза сводятся
к необходимости проведения лечения
повторными курсами (начальная суточная
доза витамина D 25 000-30 000 ЕД, максимальная
— 75 000-150 000 ЕД, доза оксидевита 0,5-1,5 мкг в
сутки), т.к.
при отмене препаратов часто
наблюдаются рецидивы (так называемые
метаболические кризы, прогрессирование
остеопороза и рахитоподобных изменений
костной ткани). В комплекс лечения
включают препараты кальция, фосфора,
витамины А, С, Е, группы В в возрастных
дозах.
Показано ограничение поваренной
соли и включение в рацион продуктов,
оказывающих ощелачивающее действие, а
также богатых калием. В фазе ремиссии
назначают массаж, соляно-хвойные ванны.
Хирургическая коррекция при болезни
де Тони — Дебре — Фанкони целесообразна
только при развитии тяжелых костных
деформаций и достижении стойкой
клинико-биохимической ремиссии в течение
2 лет.
Источник: https://studfile.net/preview/5962712/page:3/